Next-Generation Pain Control with Novel Stony Brook Fatty Acid Binding Protein (FABP) Inhibitors (SB-Fls)
Fatty Acid Binding Proteins, Endocannabinoid System and Next-Generation Pain Control
Fatty acid binding proteins (FABPs) are a family of cytosolic proteins that play a critical role in transporting fatty acids to various cellular compartments, including membranes, enzymes, and nuclear receptors. These proteins regulate lipid metabolism and signaling by modulating the intracellular availability of lipids.
One isoform of FABP, predominantly expressed in epidermal cells, is FABP5. This protein is particularly involved in endocannabinoid signaling, functioning as a transporter for anandamide (AEA), an endocannabinoid that influences pain perception through the activation of cannabinoid receptor 1 (CB1). FABP5 facilitates the metabolism of AEA via fatty acid amide hydrolase (FAAH). By regulating extracellular AEA levels, FABP5 indirectly modulates cannabinoid receptor signaling, with significant implications for pain management and anti-inflammatory pathways.
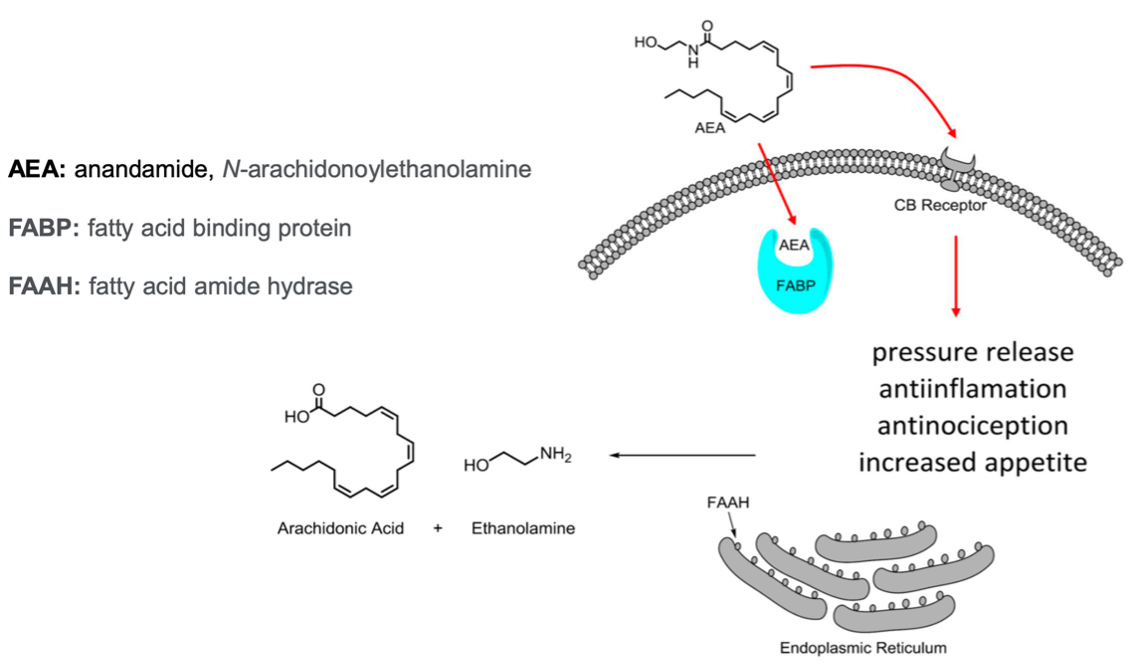
Traditional pain management therapeutics, including opioids and nonsteroidal anti-inflammatory drugs (NSAIDs), are often associated with significant limitations, such as the potential for addiction, the risk of overdose, and adverse side effects like gastrointestinal bleeding. In contrast, the inhibition of FABP5 offers a novel therapeutic approach by preventing the degradation of AEA by FAAH. This mechanism elevates AEA levels, enhancing the activation of CB1, which promotes increased pain tolerance and exerts anti-inflammatory effects.
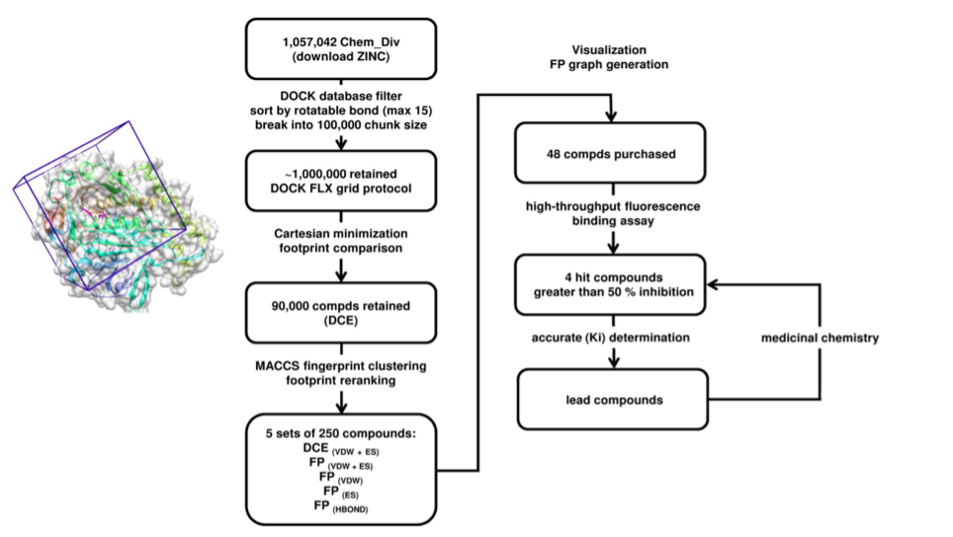
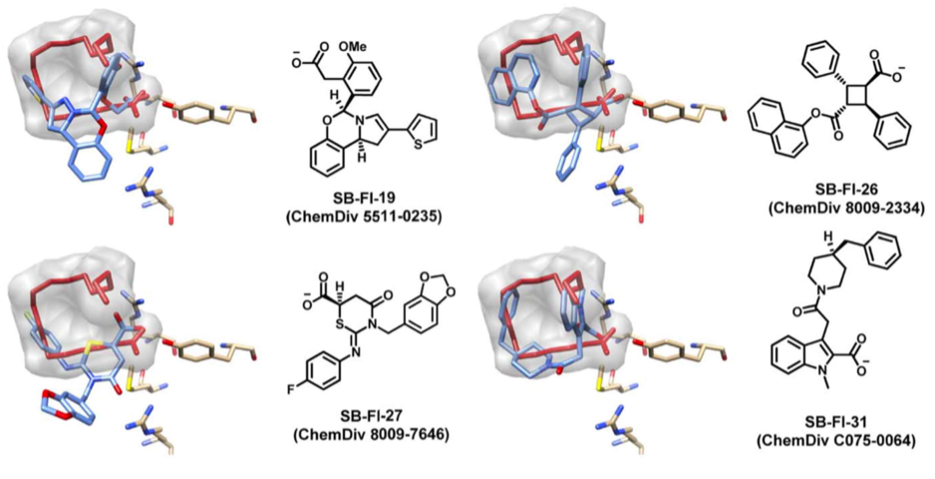
We performed a virtual screening of over one million compounds using DOCK and employed a novel footprint similarity scoring function to identify small molecules with binding profiles similar to oleic acid, a natural FABP substrate. Forty-eight compounds were purchased based on their footprint similarity scores (FPS) and assayed for biological activity against purified human FABP5, employing a fluorescent displacement-binding assay. Four compounds were found to exhibit approximately 50% inhibition or greater at 10 µM. The most potent inhibitor, -truxillic acid 1-naphthyl ester (ChemDiv 8009-2334), was determined to have Ki value of 1.19 ± 0.01 µM. It is worthy of note that α- and -truxillic acids as well as their derivatives were reported to show anti-inflammatory and anti-nociceptive effects in mice in-vivo, albeit their mechanism of action was not studied. Accordingly, a novel α-truxillic acid 1-naphthyl mono-ester (SB-FI-26) was synthesized and assayed for its inhibitory activity against FABP5, wherein SB-FI-26 exhibited strong activity (Ki 0.93 ± 0.08 µM). Additionally, SB-FI-26 was found to act as a potent anti-nociceptive agent with mild anti-inflammatory activity in-vivo (mice at 20 mg / kg i.p.), which strongly supports our hypothesis that the inhibition of FABPs and subsequent elevation of anandamide level is a promising new approach to drug discovery.
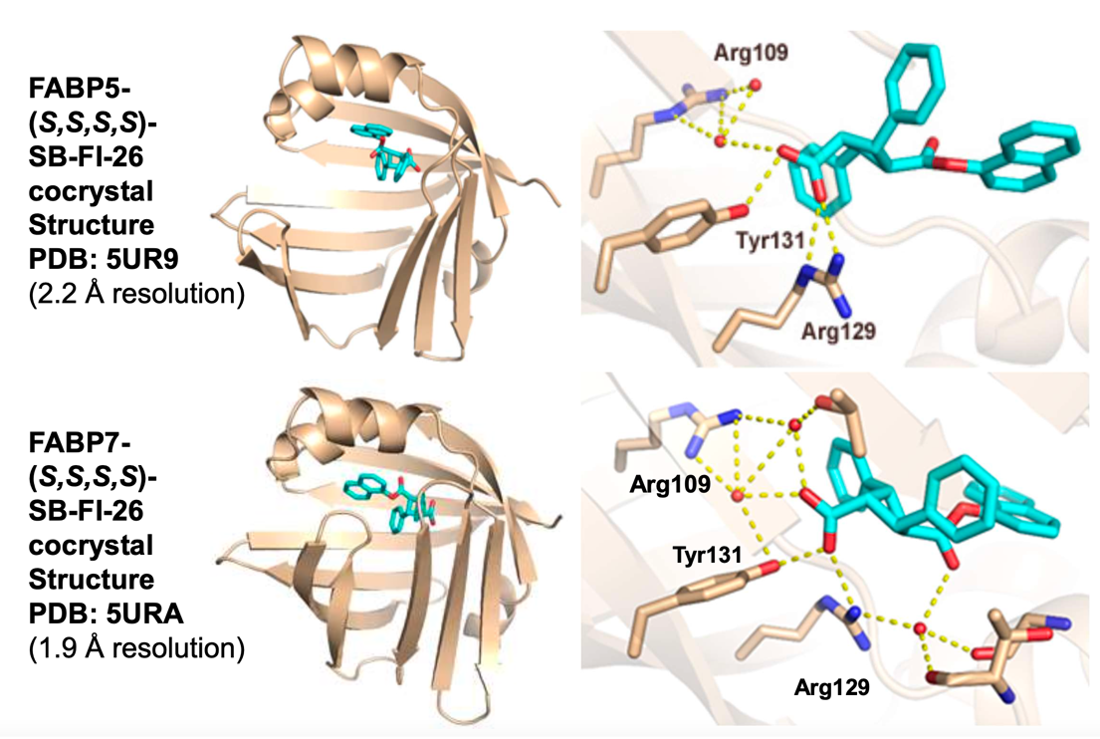
Further structural studies revealed the X-ray crystal structures of SBFI-26 bound to FABP5 and FABP7. The crystal structures for both proteins highlighted an important canonical interaction at the binding site involving Arginine and Tyrosine residues. These structural insights provide a foundation for the rational design of more potent and selective FABP inhibitors, enabling researchers to predict how structural modifications to the inhibitor could enhance binding affinity and therapeutic efficacy.
The Ojima research group conducted two extensive structure-activity relationship (SAR) studies based on the truxilic acid monoester (TAME) scaffold to further optimize bioactivity. Utilizing molecular docking and ADME/T predictions, new TAME analogs were rationally designed to enhance their therapeutic potential.
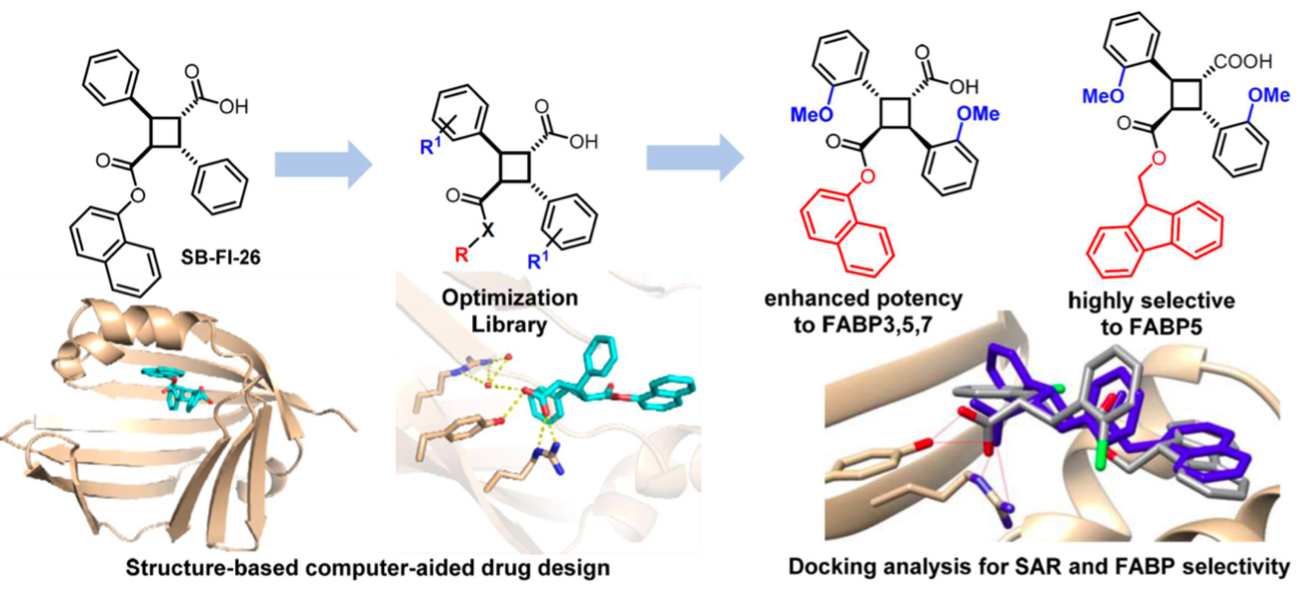
SAR studies for FABP5 inhibitors highlight the importance of targeting key residues within the FABP5 binding pocket while optimizing functional groups for increased potency, and selectivity. By systematically modifying molecular structures, researchers have developed compounds with enhanced therapeutic potential for pain management.
Chemical Synthesis of TAMEs
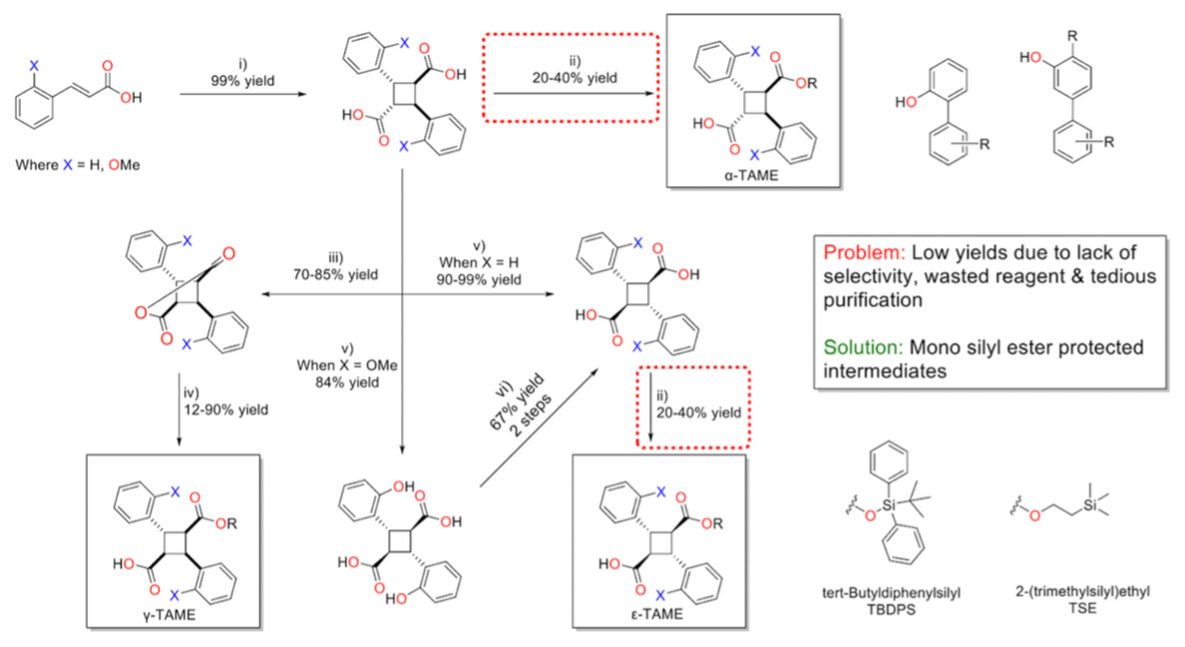
The synthesis of TAME compounds begins with trans-cinnamic acid or trans-2-methoxycinnamic acid. A [2+2] solid-state photoreaction yields the desired α-truxillic acid (α-TA) core in quantitative yields. Esterification of α-TA with dehydrating agents such as EDC, HCl produces the α-TAME, although this reaction typically results in low yields.
To synthesize γ-TAMEs, α-TA is refluxed in acetic anhydride, forming the γ-truxillic anhydride intermediate. Subsequent nucleophilic attack on the anhydride produces the γ-TAME in moderate to high yields. The ε-TA core is obtained via potassium hydroxide fusion at 350–380°C. ε-TAMEs are synthesized using a similar approach to that for α-TAMEs.
Previous SAR studies aimed at optimizing TAME compounds have focused on modifying the ester linkage of the molecule. After screening various TAMEs in silico for criteria such as docking scores, CLogP < 7.0, and ADMETox filters, the selected TAMEs were synthesized by the Ojima research group. Most of the TAMEs in this study feature a 1,2-biphenyl or 1,3-biphenyl scaffold for the ester linkage, with these phenols synthesized via Suzuki–Miyaura coupling in moderate to high yields.
Sustainable Recycling Scheme
TBDPS protection of α-truxillic acid (α-TA) yields the desired α-truxillic acid monosilyl ester (α-TAMSE) along with unreacted α-TA and the byproduct α-truxillic acid disilyl ester (α-TADSE). By employing a 2:1 molar ratio of α-TA to TBDPS-Cl, α-TAMSE can be isolated in yields of up to 75%. Excess α-TA can be recovered through filtration, while α-TADSE can be recycled back into α-TA via acid hydrolysis. This approach enables 100% material recovery or utilization, facilitating the synthesis of α-TAMEs with improved yields, simplified purification processes, and enhanced sustainability.
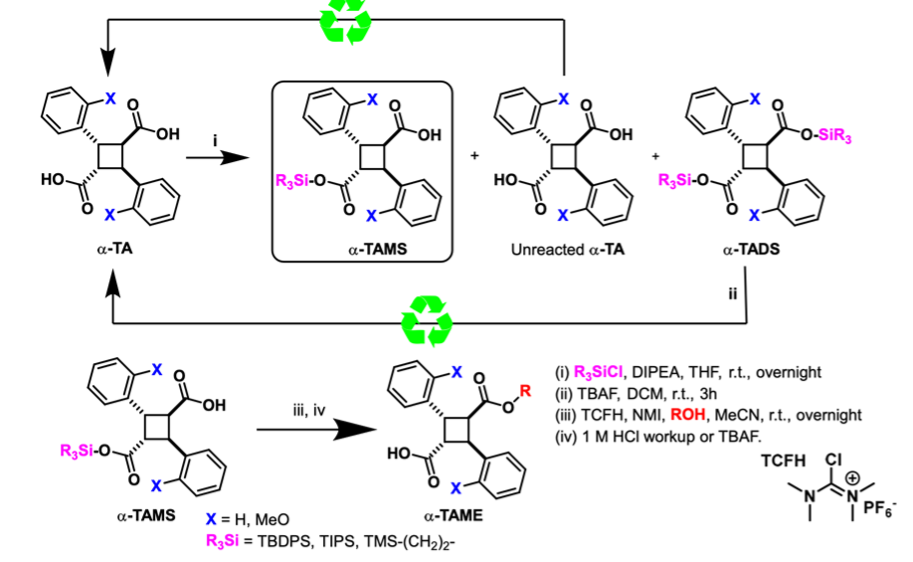
α-Truxillic acid (α-TA) and α-2-methoxy-truxillic acid (α-MeO-TA) were synthesized through [2 + 2] photocycloaddition of E-cinnamic acid and E-2-methoxycinnamic acid, respectively. Hydroxy-1,1′-biphenyl derivatives were prepared via Suzuki coupling from aryl halides and arylboronic acids/esters unless commercially available.
The synthesis of α-TAMEs involved the reaction of diacid chlorides—generated in situ from α-TA or α-MeO-TA with thionyl chloride, or via condensation with hydroxyarenes or alcohols using EDC as a dehydrating agent. Similarly, γ-TAMEs were synthesized by reacting γ-truxillic anhydride (γ-TAA), obtained through the epimerization-cyclization of α-truxillic acids, with hydroxyarenes or alcohols in the presence of a base.
Furthermore, ε-truxillic acid (ε-TA) was prepared from α-TA via alkali fusion at 325 °C and converted to ε-TAMEs using methods analogous to those employed for α-TAMEs.
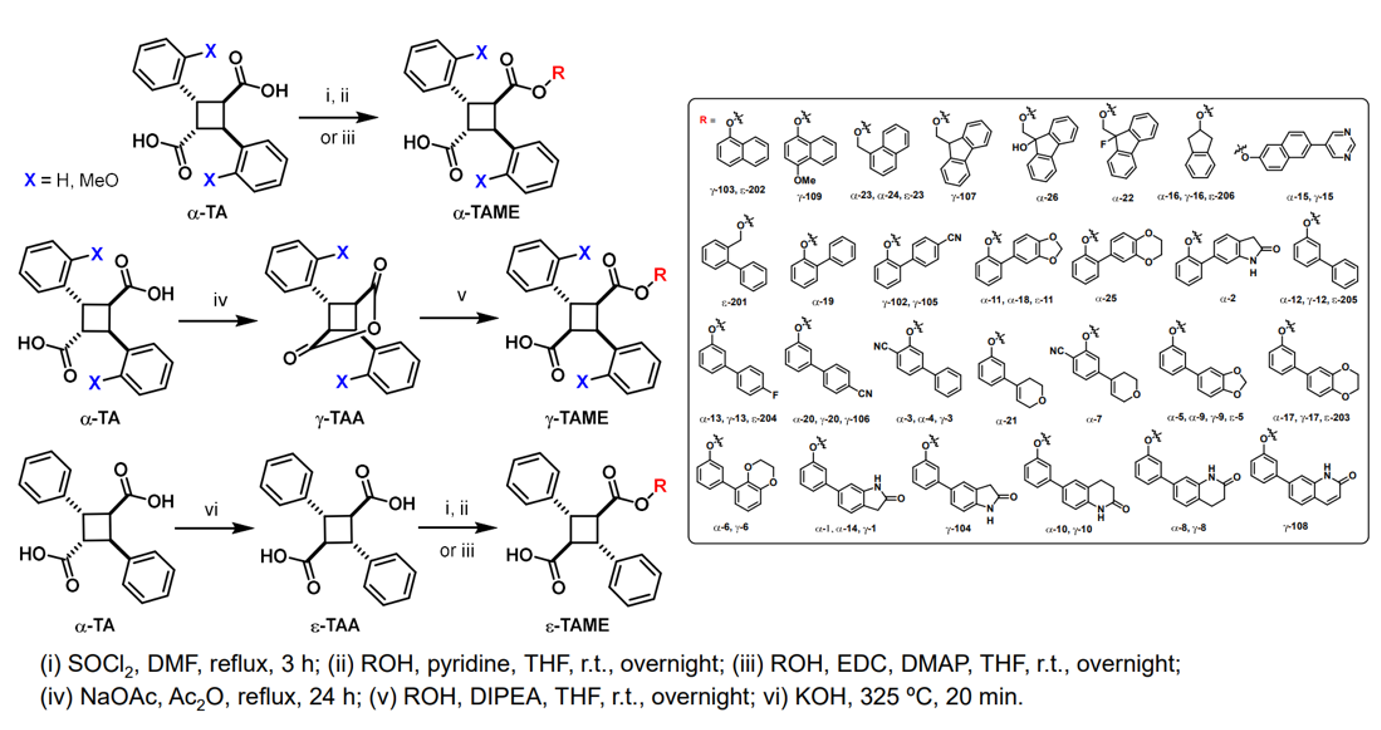
SAR Study on Novel Truxillic Acid Monoester -Based Inhibitors of Fatty Acid Binding Proteins as Next-Generation Antinociceptive Agents: 3rd-Generation Stony Brook FABP Inhibitors (SB-FIs)
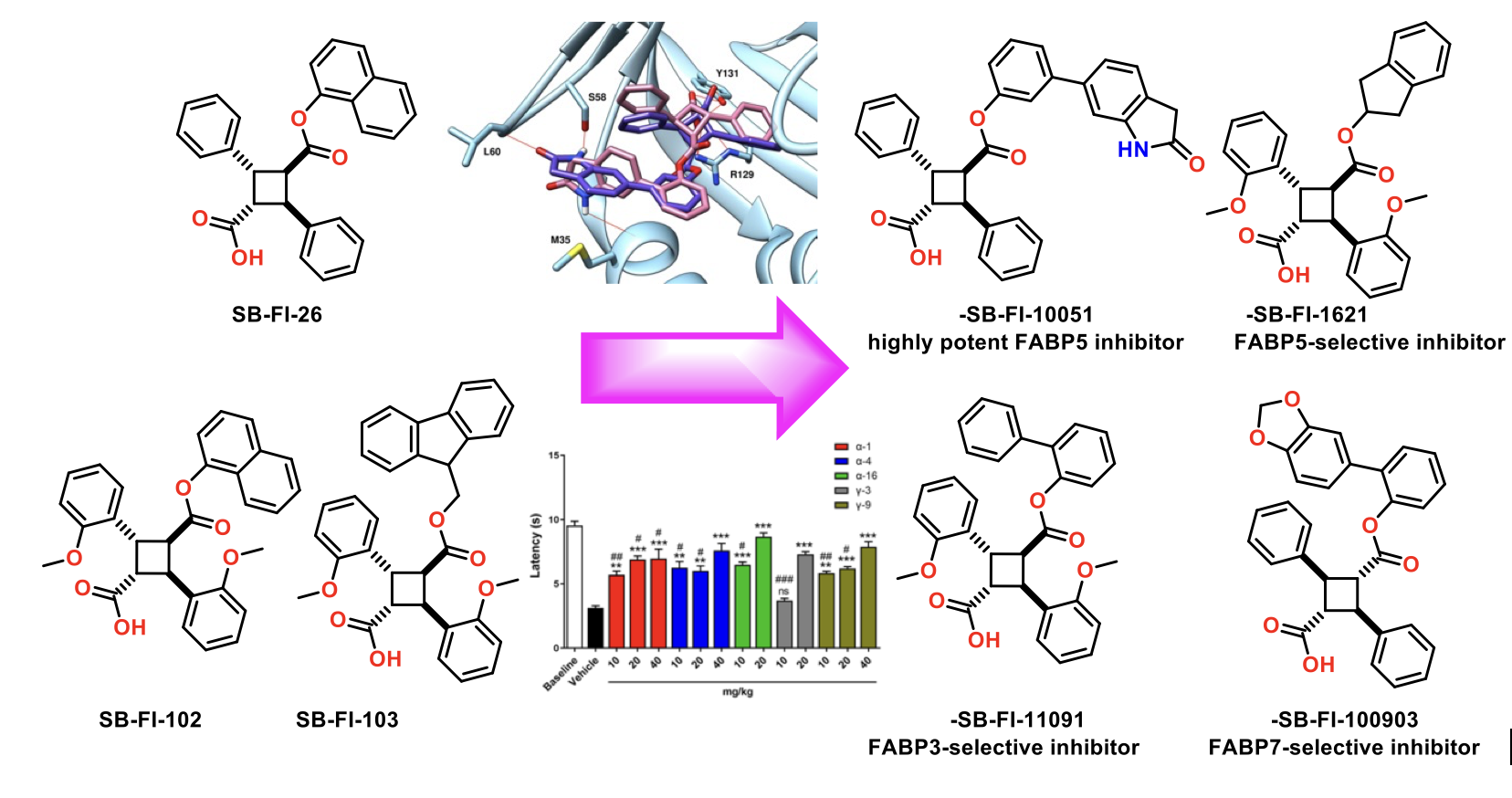
The design and discovery of highly potent and FABP5-selective truxillic acid (TA) monoesters (TAMEs) is the primary aim of this study. On the basis of molecular docking analysis, ca. 2,000 TAMEs were designed and screened in silico, to funnel down to 55 new TAMEs, which were synthesized and assayed for their affinity (Ki) to FABP5, 3 and 7. The SAR study revealed that the introduction of H-bond acceptors to the far end of the 1,1’-biphenyl-3-yl and 1,1’-biphenyl-2-yl ester moieties improved the affinity of a-TAMEs to FABP5. Compound g-3 is the first g-TAME, demonstrating a high affinity to FABP5 and competing with a-TAMEs. We identified the best 20 TAMEs based on the FABP5/3 selectivity index. The clear front runner is α-16, bearing a 2-indanyl ester moiety. In sharp contrast, no e-TAMEs made the top 20 in this list. However, α-19 and ε-202, have been identified as potent FABP3-selective inhibitors for applications related to their possible use in the protection of cardiac myocytes and the reduction of α-synuclein accumulation in Parkinson’s disease. Among the best 20 TAMEs selected based on the affinity to FABP7, 13 out of 20 TAMEs were found to be FABP7-selective, with α-21 as the most selective. This study identified several TAMEs as FABP7-selective inhibitors, which would have potentially beneficial therapeutic effects in diseases such as Down’s syndrome, schizophrenia, breast cancer, and astrocytoma. The molecular docking analysis has provided rational explanations for the observed binding affinity and selectivity of the FABP3, 5 and 7 inhibitors, including their a, g and e isomers, in this study.
ART26.12 (SBFI-1621) Advances to Clinical Trial Phase I for the Treatment of Chemotherapy-Induced Neuropathy
Following a partnership with Artelo Biosciences, Inc., a clinical-stage pharmaceutical company, SBFI-1621 (designated as ART26.12) has received FDA clearance to proceed with first-in-human clinical trials. The initial focus of these trials is chemotherapy-induced peripheral neuropathy (CIPN). Peripheral neuropathy refers to a condition in which there is damage to the peripheral nerves. These nerves are responsible for transmitting signals between the central nervous system and the rest of the body. Chemotherapy-induced peripheral neuropathy (CIPN) is a common and often painfully debilitating complication of cancer therapies, sometimes resulting in reduction or cessation of treatment. No currently approved treatment exists for CIPN. Diabetic neuropathy refers to a type of nerve damage that occurs as a complication of diabetes. It is caused by long-term high blood sugar levels, which can lead to damage of the blood vessels and nerves throughout the body. The prevalence of diabetic neuropathy is significant due to the increasing number of people with diabetes worldwide.
Other Indications of Stony Brook FABP5 Inhibitors
Preclinical studies of ART26.12 (SB-FI-1621) and other SB-FIs demonstrated significant pain relief in a rodent model of osteoarthritis (OA), outperforming naproxen, a commonly used NSAID. The compound showed improved weight-bearing on affected limbs and exhibited a clear dose-response relationship. This novel, peripherally acting, non-opioid, and non-steroidal analgesic targets fatty acid binding protein 5 (FABP5), which is linked to abnormal lipid signaling in various diseases.